Maybe before you start your lab how about some free coffee? check
this out
Introduction and Purpose
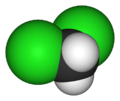
The purpose of this lab was to extract caffeine from tea. The molecular structure of the of caffeine the molecule of interest for the extraction, caffeine, is shown below.
Facts about Caffeine (maybe even fun facts):
In its pure form its a white substance that melts at 236 degrees Celsius. It is an irritant and is considered toxic. Tea is said to be about 3% caffeine by weight, but it ranges across teas.
Caffeine can be found in coffee, energy drinks, tea (clearly, hence its used in lab), No doz, Midol and more!
Techniques used:
Vacuum Filtration, use of micro pipettes funnel, centrifugation and use of drying agents
Procedure
The procedure used for the lab varied from the exact directions listed in the Lab Manual. This section will provide the procedures carried out, including the alterations from the manual. Aside from centrifugation the procedures were all carried out under a hood, all individuals involved wore
nitrile gloves and safety goggles. Glassware was cleaned, rinsed with deionzed water and if necessary acetone before usage.
First the contents of 2 tea bags were put into a 150mL beaker. The mass of the tea was about 4.681 grams. Then about 2.50 grams of Calcium Carbonate were added followed by . 50mL water.
The mixture was boiled on a hot plate on a medium heat level, during this time period a watch glass was placed on the top of the beaker. After 10 minutes of boiling the mix was set on the bench top to cool so that it was warm to the touch.
During the cooling period a 5-cm Buchner funnel was put inside a 125mL filter flask with an arm that could be connected with proper tubing to a vacuum. The funnel itself had a piece of Whatman No. 54 filter paper inside that had been wetted slightly. Additionally a clean 150mL beaker was filled with 12mL water (measured with a graduated cylinder) and the 12mL level was marked with a piece of tape, the water was poured out. While no picture was taken of the lab set up the image below shows the set up despite the use of an adapter as shown in the image.

The remaining solution was put through the filter and vacuum apparatus that was assembled during the cooling period. The vacuum was turned on and was allowed to run until filtered solution stopped coming out of the funnel tip and a pool of solution sat at the bottom of the flask (indicating that all the sample had gone through). The filtered sample (from the flask) went into the beaker that had the 12mL mark.

The sample was boiled shortly using a hot plate till it was reduced to the 12mL mark. The sample was then allowed to cool on the bench top till it was warm to the touch.

The sample was transferred to a 15mL centrifuge tube with a screwcap, 2mL of
dichloromethane were added to the solution (dichloromethane is also known as Cl2CH
2).
The solution was shook for about a minute and put in the centrifuge for about a minute. Upon completion of centrifugation 2 layers of material became apparent. A brown layer said to contain
tannins and other materials and a green layer at the bottom containing the
caffeine dissolved in the dichloromethane. A pipette was used to extract the green organic layer, which was placed in another centrifuge tube. Another 2mL of dichloromethane were added to the solution, the tube was capped, solution shook, centrifuged and the portion of the solution containing the caffeine was extracted with the pipette again. The process of adding 2mL dichloromethane, putting the cap on the tube, shaking, centrifugation and removal of the green layer was conducted a total of 4 times. After the 3rd centrifugation the layers did not separate in a distinct fashion, a small amount of
sodium chloride (NaCl) was added to aid the process. The layers separated in a more distinctively, the organic layer was removed then the final set of preparation, centrifugation and extraction was completed.
Next
anhydrous magnesium sulfate (a drying agent) was added to the tube with extracted dichloromethane and caffeine solution. This mix was allowed to sit for about 5 minutes with occasional swirling till the mix became clear. Then a microfunnel was prepared by tightly packing small piece of cotton at the tip of the pipette where it began to taper. It was clamped to a vertical bar in the hood such that it was about half way into a 25mL Erlenmeyer flask.
The dichloromethane and caffeine solution were transferred to the micropipette via a clean pipette and was allowed to go through the microfunnel and go into the clean 25mL Erlenmeyer flask. The remaining magnesium sulfate in the centrifuge tube was rinsed with .5mL dichloromethane so all contents could go through the microfunnel.
 |
| Above are magnified caffeine crystals |
The 25mL Erlenmeyer flask containing the filtered solution was placed on a heat plate on a relatively low heating level, allowing for the dichloromethane to evaporate. The flask was removed once a dry dark green residue formed at the bottom of the flask and all the dichloromethane seemed to have evaporated. The flask was allowed to cool on the bench top. Then the sample was weighed. The percent recovery was then calculated by dividing the recovered mass of caffeine by the initial amount. Once everything was completed a cork was placed tightly on the flask and the sample was stored to be used for another lab experiment. For clean up the tea was thrown in the trash being that they are not hazardous. Materials remaining from the extraction were put in the non hazardous waste bin. Meanwhile the centrifuge tube that previously had dichloromethane was left on its side to allow all dichloromethane to evaporate fully and then was rinsed. All glassware were cleaned with soap and water, dried, and given a quick rinse with deionized water.
Questions (Feel free to ask more, heck I'll even try to answer them for free!)
Why was the tea boiled (in water)? Turns out caffeine is actually highly soluble in hot water and it turns out boiling the tea leaves in hot water allows for the caffeine to be released. Hence we are able to extract the caffeine from the water later in the procedure.
Why is the aqueous tea
solution cooled to 15-20°C before the
dichloromethane is added?
The boiling point of CH2Cl2 is 40C water boils higher than this. If dichloromethane gets too hot, it emits highly toxic fumes of phosgene. Now would be a great time to say that dichloromethane is actually kind of dangerous and acts as an irritant to the respiratory system (that thing you breath with). If heated to decomposition in flame or hot surface to form toxic gas
phosgene and corrosive mists of hydrochloric acid are also formed. AND YOU GET TO BREATH IT IN!!!!!! YAYYYYY!!!!! To quote MSDS " Continued exposure may cause increased light-headedness, staggering,
unconsciousness, and even death. Exposure may make the symptoms of
angina (chest pains) worse"
GO HERE TO FIND OUT MORE Oh yeah and I forgot its probably carcinogenic too....keep it cool and keep safe.
Now you may ask what is MSDS? They are Material Safety and Data Sheets. Home page is
http://www.msds.com/ go there to learn stuff about stuff.
Why is the tea solution cooled before dichloromethane is added? Think of adding sugar or salt to water, water has higher solubility levels for many compounds at higher temperatures that can even allow for super saturation. But while it cools the solubility and the ability to hold the caffeine decreases as the temperature drops. Slowly but surely the caffeine becomes undissolved and then we add the dichloromethane to add the process of extraction.
How do I calculate percent recovery of the extraction of caffeine from the tea leaves?
Percent Recovery = amount recovered = Insert mass of what you got x 100% = % recovery
Initial amount * what it should have been
* Note as stated earlier tea is 2-5 % caffeine by weight, so unless you know the exact number multiply the mass of how much tea you started with by 3.5% to see the total caffeine you could have been able to extract. Don't be surprised if this number is rather low.
Why does adding salt (NaCl) to the aqueous layer sometimes help break up emulsions that may form in an extraction?
Hmmm...not sure of this one but here is my guess.
NaCl is pretty soluble in water. And emulsions by definition are non polar substances surrounded by polar substances. Fun fact mayonnaise is an emulsion (True life I'm a food network dork). The NaCl will attract these water molecules deterring them from dealing with the emulsion. It helps provide a stronger inter phase between solutions too! Also adding salt can cause salting out.
In the event you used 1-Propanol and Salt
NaCl is used as a salting out technique to increase the dielectric constant of the water layer. By doing so, 1-propanol will separate out from the salt water and this allows the extraction of caffeine into the 1-propanol layer. In order for the 1-propanol to separate out from the water layer, the water must be saturated with NaCl. This can only be achieved if NaCl is added in excess amount.
When salt is added to the water layer, it decreases the solubility of caffeine. This is due to the fact that caffeine is only slightly polar in nature. Thus, the solubility equilibrium will now shift to the 1-propanol. This will cause the partition coefficient to decrease.
In the event you added Calcium Hydroxide. Here is why.
Calcium hydroxide helps to precipitate out the tannic acid as calcium tannate in tea leaves. If
sodium hydroxide is used instead, no precipitate will form (sodium tannate is soluble in water). The caffeine crude product will be contaminated with the tannate salt. And that would be a annoying
Caffeine is a white powder, why the hell is my sample GREEN!!!!?????
The sample is green because well lets face it you extracted it from tea...Tea is a plant. Plants have
chlorophyll. Chlorophyll is green. Turns out you probably have chlorophyll in your sample. Tune in some other time and maybe I'll have a way...or do a lab that finds a way to eliminate and purify the extracted caffeine.
Why do we centrifuge in the caffeine extraction?
You will use a centrifuge in this experiment break up emulsions once they form, separating the aqueous and organic liquid emulsion.
Now you may ask what is an emulsion? An emulsion is a suspension of one liquid as droplets in another (the two liquids must be insoluble in one another). Think water in oil or oil in water they don't really mix. To
avoid them, you can shake mixtures of insoluble liquids gently and add
salt to aqueous layers remember that salting out thing you did by adding NaCl.
Meanwhile if you're bored feel free to visit and join.
 What is the role of sodium carbonate in the extraction of caffeine in tea leaves? and also what is the principle involved in extraction?
What is the role of sodium carbonate in the extraction of caffeine in tea leaves? and also what is the principle involved in extraction?
The
sodium carbonate acts as a base - you could use
sodium hydroxide
instead. When you boil tea leaves tannins dissolve in the water as well
as the caffeine. If you do not use a base the tannins will also be
extracted into the solvent (i.e.
methylene chloride)
used in the subsequent extraction . The base converts the tannins into
their sodium salts - being ionic these salts are not soluble in solvents
like methylene chloride so remain in the aqueous layer during
extraction. This allows purer caffeine to be extracted.
























{kind=link}
{kind=link}
{kind=link}