So this isn't the best way to make alcohol I guess but still never the less its alcohol...lets make some. Specifically we will be synthesizing ethanol by fermenting
sucrose with the help of some yeast. Ethanol is also known as Absolute alcohol, Alcohol, Drinking alcohol
Ethyl alcohol, Ethyl hydrate, Ethyl hydroxide, Ethylic alcohol, Ethylol, Grain alcohol, Hydroxyethane, Methylcarbino.
Go here to learn about ethanol.


General Information:
What we are going to talk about.
C2H22O11 plus H2O breaks into 2 C6H12O6 plus zymase gives us 4 CH3CH2OH (ethanol) + 4 CO2
Place 40 mg sucrose in a 500 mL Erlenmeyer, add 200 mL water and 3.0 g dry yeast. Stire till sugar dissolves and you can't really see the yeast.
Add 35 mL Pasteurs salt - stir to mix. Close the flask with a stopper fitted witha piece of bent glass tubing.
Fill a test tube halfway with a saturated Ca(OH)2 (also known as limewater) - submerge other end of glass tube into test tube so its about 1 cm below surface of the solution. Store for a week.
After a week come back and filter the solution via vacuum filtration in a 500 mL filter flask. Rinse flask with water. Now take the filtrate in the Erlenmeyer and place in a round bottom flask, add 2 boiling stones and assemble for simple distillation. Set so the Alcohol flows into the recieving flask at 1 drop/sec. Heat till 50 mL are collected.
To find the density of this weight a tared 10.0 mL erlenmeyer, add 10.0 mL of the distillate and mass/volume = density which corresponds with a percent alchol by volume.
Take the sample (all of the simple fractional distilation sample) and prepare for fractional distillation. Use a 100 mL round bottom flask, add some boiling stones. In the Fractional distillation either pour some glass beads or stainless steel sponge. Turn heat to moderate amount and wait. Collect 3 different samples each called Fraction 1, 2, 3 etc.
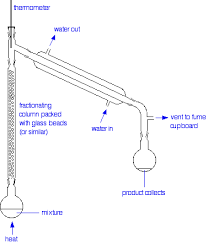
Fraction one should be the first 10 mL sample, Fraction 2 is the sample from 78 to 80 degrees Celcius and Fraction 3 is really rubish.
To determine the percent alcohol by volume or by mass use the same technique as above. Take a known volume, find its mass. Mass divided by volume = density. Use tables or online literature valus to find the percent alcohol by volume by the corresponding density. Remember water is more dense than alcohol. Hence water sits below alcohol if you pout them together. Ideally you would want your sample to weigh less.
Questions
What does the density and percent composition results tell you about the relative efficiencies of simple vs. fractional distillation? In other words, the distillation efficiency is higher when the distillate density is _________ and percent ethanol is ______________.
Blank 1) lower Blank 2) higher
Distillate from simple distillation does not have as high of percent alcohol as those of fractional distillation, hence fractional distillation has a higher efficacy. When distillation efficacy is higher the density of the solution is lower and its alcohol content is greater.
2. Which method gives purer ethanol? Briefly explain reasons for the difference in efficiencies of these two distillation methods, assuming no large technical errors were made.
Fractional distillation should result in purer ethanol. The difference is in part due to difference in apparatus, which provides more surface area to allow for cooling and condensation of the sample, the increase in plates and allow to get closer to the azeotrope.
3. How does the recorded temperature relate to the composition of the distillate?
The lower the boiling point of the solution the greater the likelihood that there is more ethanol in the solution. Ethanol has a lower boiling point than water, hence it is more volatile. Temperature and composition are directly related because the boiling point is contingent upon the compsition of the solution and the more volatile substances boil first.
4)What is an "Azeotrope"?
Its where you have liquid and vapor with same boiling point so no more enrichment can occur via distillation.
5) What makes fractional distillatation more efficient that simple distillation in terms of the aparatus. Being that in this case the two liquids have a similar boiling point.
The distillation column makes fractional distillation more efficient. Its filled with glass beads or steel wool. This elongated column allows for more surface area fr vapor to become cool and in liquid form again. Then it heats up again. Allowing for more plates (cycles of warming to vapor and being cooled again).
In each plate (heating till vaporazation till cooling and retuning to liquid form via condensation) in falls down back the fractional distillation tube the percent alcohol actually increases till the azeotrope is reached because both liquids have simlar boiling points they go through the same states.
Check out this image it may help.
Anyways if you are bored and or broke as always go here:






























{kind=link}
{kind=link}
{kind=link}